
What
is small RNA (sRNA)?
50-400bp RNA molecules, abundant in bacteria, not translated to
protein, but function directly as structural, regulatory or even catalytic RNA.
Involved in:
![]() Transcriptional
regulation
Transcriptional
regulation
![]() RNA
processing and modification
RNA
processing and modification
![]() Messenger
RNA stability and translocation
Messenger
RNA stability and translocation
![]() Translation
regulation
Translation
regulation
RNA molecules may form secondary structures stabilized by base-pairing
of two different parts of the molecule.
What
do we know about sRNA?
![]() First
few were discovered by chance, and found to have regulatory function
First
few were discovered by chance, and found to have regulatory function
![]() In
recent years computational approaches were employed to predict new sRNAs
In
recent years computational approaches were employed to predict new sRNAs
![]() During
2001, several works were published and 55 new sRNAs were predicted and
identified
During
2001, several works were published and 55 new sRNAs were predicted and
identified
Regulation mechanisms:
![]() Binding
to complementary sequences by basepairing (--> SRTF's
focus)
Binding
to complementary sequences by basepairing (--> SRTF's
focus)
![]() Interacting
with proteins
Interacting
with proteins
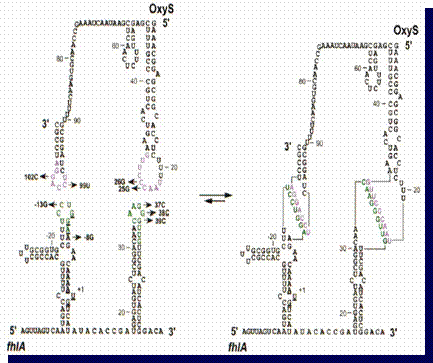
One of the known examples of
sRNA regulation includes a sRNA molecule called oxyS which
regulates the translation of a gene called fhlA in response to
oxidative stress in E. Coli. Both oxyS and fhlA
molecules fold independently to form secondary structures of stems and loops.
When the two molecules are present in the cell at the same time, oxyS inhibits
the translation of the fhlA gene by forming 2 links (loop to loop antisense
basepairing) between the two molecules, capturing fhlA's
Shine-Dalgarno and AUG region (which are vital for translation initiation).
What
is SRTF?
SRTF stands for Small RNA Target Finder.
It is a heuristic program, written in Perl, which grades all E. Coli genes by their relative quality as target genes for
a given sRNA sequence. The program uses different parameters to score each
gene, including free energy calculations, secondary structure of the given sRNA
sequence and analysis of sRNA-target gene match pattern.
Because there are only few examples of translation regulation by
antisense basepairing of sRNA, SRTF uses a heuristic approach, trying to find
genes that best answer several criteria which characterizes the known examples.
SRTF scans each gene for basepairing matches to the given sRNA, then it grades
every match based on how well it can influence translation initiation (based on
parameters like match length, match proximity to the gene translation
initiation site, etc.). The final analysis phase of every gene,
includes the detection of match combinations, that can jointly maximize the
chances of the gene translation prevention by the given sRNA.
Program Input:
![]() sRNA
annotation
sRNA
annotation
![]() The
complete E. coli genome file including annotations, downloaded from NCBI.
The
complete E. coli genome file including annotations, downloaded from NCBI.
![]() MFold
output files
MFold
output files
1.
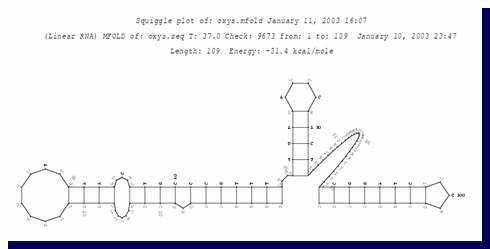
Collecting sRNA secondary structure data
The MFold program (Zuker M. 1989) predicts the secondary structure of a
given RNA molecule based on free energy considerations. SRTF uses MFold in
order to get the predicted secondary structure of the given sRNA sequence. SRTF processes the MFold output in order to
get the following information about the sRNA structure:
Basepairing –
Specifies whether a certain position in the sRNA sequence is basepaired to a
different base of the molecule, or whether it is unbasepaired (free, and thus
more easily available to form basepairing with bases of the target molecule).
Extension –
Specifies the "height" of a given base in the sRNA molecule. The
extension value of a given sRNA position is calculated as its distance (in bp) from the beginning of the stem it is located on. All
bases located in a loop will have the same height – the length of the stem
leading to that loop. SRTF assumes that sRNA regions which are located on a
long extension would be more readily available to reach key regions on the
target gene and attach to them, then sRNA regions buried deep within the
molecule secondary structure.
minExtensionHeight
- Minimal height of sRNA window to be considered as extension.
maxExtensionHeight - Maximal height of sRNA window to be considered as
extension.
Picture: An example of MFOLD graphic
output for the sRNA oxyS.
1.
Scanning the genome for hits
SRTF scans all genes, looking for matches of a gene window to a sRNA
window. SRTF currently uses a 5 base minimal window size. Every match found (at
least 5 sRNA bases which are complement to same number of bases in the currently
scanned gene) is being stored for later analysis. When this phase is completed,
SRTF holds a list of hits found within each gene, when every hits
includes its 4 basic properties (raw properties): sRNA position, target
position, hit length and hit free energy.
Hit (Match) – Sequence of X bases in a gene, complementary to a
sequence of X bases in the sRNA. Every hit contains 4 properties:
|
HIT = (sRNA pos , target pos , length ,energy) |
sRNA Position – Position on the sRNA sequence, on which the match
begins.
Thanks to phase 1 (where SRTF collected data about the sRNA secondary
structure), once we have the sRNA position of a
certain hit, we know if the hit is located on a basepaired region or on an extension
of the sRNA molecule. That information enables us to evaluate the quality of
the hit and grade it accordingly in the next phase.
Important Note: SRTF displays sRNA positions starting from the 3`.
This way it's easier to immediately see the relative position of two or more
hits. Therefore, base number "sRNA pos" (starting from 3`) on the
sRNA sequence is complement to base number "Target pos" on the target
gene (starting from 5`).
Target Position (mRNA position)
– Target gene position on which the
match begins. Refers to the currently graded gene – Actually it's not a target
yet but only a target candidate.
Hits whose "Target Position" located around the
Shine-Dalgarno & AUG region of the target gene are more likely to influence
the translation initiation of that gene, making them interesting to us as we
are interested translation regulation.
Important Note: Target Position 0 is the first base of the AUG on
the target gene.
Hit Length – Number of bases included in the match. SRTF has a
minimal match length of 5. Every hit includes the exact same number of bases on
the sRNA molecule and on the Target gene mRNA molecule.
Hit Energy – For each match, SRTF calculates the free energy
based on basepairing and stacking energy (Turner
D.).
Hits which contain many G and C bases would usually have a much lower
energy value (The lower the value – the stronger the match is).
Hits with a very negative value of free energy would mean that the hit
is stronger – more energy is needed to break that bond. Strong matches are
preferable energetically, thus more likely to occur.
analyzeHitsFromPos – This SRTF parameter
defines mRNA position lower bound of hits that shall be included in the gene
grading process. Only hits which their target position is higher than that
parameter will be included.
analyzeHitsUntilPos – This SRTF parameter
defines mRNA position upper bound of hits that shall be included in the gene
grading process. Only hits which their target position is lower than that
parameter will be included.
Note: Phases 3 & 4 are performed on all E. Coli genes sequentially. Gene after gene, SRTF first
grades each hit found on that gene independently, and then SRTF gives a total
grade for the gene by finding best hits combination and giving a score to that
combination.
In this phase, SRTF gives an independent score to each hit it
found on the currently graded gene. Hit score is calculated based on the 4
basic hit properties: sRNA position, Target position, Length and Energy.
Hit Score – Hit score specifies the relative estimated hit
quality as involved in an effective translation regulation of a gene by a given
sRNA.
Long and energetically stable matches, located on extended sRNA loops
and in close proximity to the gene's Ribosomal binding site, should get a high
score because they have a higher chance to be involved in translation
regulation. Therefore, Hit Scores are calculated based on 5 sub-scores: 2 of
them are basic hit properties (length and free energy) and the other 3 are
functions – positionScore (translates Target gene
position to Target Position sub-score), bpScore
(translates sRNA position to basepairing sub-score) and extensionScore
(translates sRNA position to extension sub-score).
|
Hit Score
= (length / lengthThreshold ) * l00 * lengthWeight + (energy / energyThreshold ) * 100 * energyWeight + (positionScore / positionThreshold
) * 100 * positionWeight + (extensionScore / extensionThreshold ) * 100 * extensionWeight
+ (bpScore / bpThreshold)
* 100 * bpWeight |
There are 3 sub-score functions included in the hit scoring process:
1. Target gene position score (Position
score) – As we are interested in hits
which can efficiently prevent gene translation, we would give precedence to
hits falling on the translation initiation region of genes. We would usually be
interested in hits which fall on the Shine-Dalgarno or on the AUG of the gene,
blocking access to the ribosome and avoiding it from translating the gene. We
would also be interested in hits which are located in close proximity to that
important region (the oxyS-fhlA example includes 2 hits on target positions -15
and 33 which capture the AUG of the target).
Hence, the Target position score function may seem like the following
graph (it depends on the parameters set) – giving the highest grade to hits
which fall on the "important region" between the Shine Dalgarno to
the AUG, a rather good grade to hits located in close proximity to the AUG
downstream, and low grades to distant hits downstream.
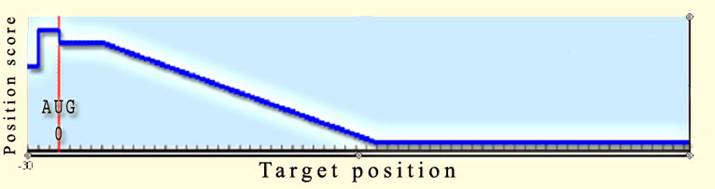
There are 7 parameters, defining the position score function:
negMark
- Upstrean
mRNA position mark which is used to score hits by mRNA position
posMark1 - First
downstream mRNA position mark which is used to score hits by mRNA position
posMark2 - First
downstream mRNA position mark which is used to score hits by mRNA position
preNegMarkScore -
Grade which will be given to hits which their mRNA position falls upstream to negMark
postNegMarkScore
- Grade which will be given to hits which their mRNA position falls between negMark and 0 (AUG)
prePosMark1 -
Grade which will be given to hits which their mRNA position falls between 0
(AUG) and posMark1
postPosMark2 -
Grade which will be given to hits which their mRNA position falls upstream to
posMark2

2. bpScore
– Basepairing Score is given to a sRNA
window and it specifies the number of un-basepaired bases within that window.
This function uses the secondary structure that SRTF got from the MFold
program on phase 1. Hits falling on un-basepaired regions in the sRNA should
get higher grade because they are more readily available to connect to
complementary window on a target gene, as the sRNA-target basepairing doesn't
have to "compete" with internal sRNA basepairing.
sRNA
windows containing consecutive un-basepaired bases will get higher bpScore than windows containing same number of un-basepaired
bases, but in a non-consecutive pattern.
3. extensionScore –
Extension score of a given sRNA window measures the height of that window on
the sRNA molecule two dimensional topography. sRNA
windows located on linker regions will get an Extension Score of 0, while
windows located within extended loops will get the height of that loop (number
of bases in the loop's stem) as the extension score.
The extension score is supposed to reflect the sRNA topology, giving
precedence to hits located on extended branches of the sRNA molecule – assuming
that sRNA extension located windows will be able to reach their complementary
target gene more easily (On the oxyS-fhlA example, both hits are located on
sRNA extensions).
Hit Scoring Weights – Assigning scoring weight to each of the hit
sub-scores enables us to control how dominant each hit property would be in the
total score. Setting a sub-score weight to 0 would actually make SRTF to ignore that property when
scoring hits.
There are 5 weights – one for each of the 5 sub-scores composing the
total hit score: positionWeight (=Target Gene Position), extensionWeight, bpWeight, lengthWeight and energyWeight.
Thresholds –
Every hit sub-score is normalized by division by a threshold value. Lower
threshold values will increase the sub-score influence on the total hit score.
There are 5 threshold parameters: lengthThreshold, positionThreshold, energyThreshold,
bpThreshold and extensionThreshold.
After scoring each hit found within the currently graded gene
independently, this phase includes the calculation of a total gene score. SRTF
tries to identify the best hits combination, so it generates all hit pairs,
scores each pair, and then sets the total gene score to be the maximum over all
pair scores and all single hit scores.
|
Gm=
max ( Sij , Si) i=1..K , j=1..K K = number of hits found on gene m Si = Score of
single hit j Sij = Score
of hits pair (i,j) |
Hits Pair Score is calculated as the sum of the two hit score, in
addition to 3 per-pair penalties/bonuses (Distance
penalty, Ratio Bonus and Cover Fraction Bonus).
|
Sij = Si + Sj – DistancePenalty
+ RatioBonus + CoverFractionBonus |
1. Distance
penalty –
Pair Distance is calculated as difference between target positions of two hits.
Distance penalty is reduced from pair score when the distance on the target
gene, between two hits, exceeds a certain value. The idea is to give precedence
to hit pairs in which the two hits are rather close one to the other on the
target gene, as we expect two tight hits to be more stable than 2 hits,
hundreds of bases apart (2 tight hits attach the target gene locally –
stabilizing each other, while allowing most of the mRNA molecule to retain it's
original secondary structure).
distancePenaltyFromPos – Defines the minimal hits mRNA distance which will trigger Distance
penalty reduction.
distancePenaltyPerBase - Number of points reduced per base. Defines how strict the Distance
penalty would be for pairs exceeding the minimal hits mRNA distance.
maxDistancePenalty
– Maximal distance penalty value. Distance penalty will not top this value.
2. Cover Fraction
Bonus - As we are interested in hit pairs which avoid the ribosome
from accessing the translation initiation site of the target gene, we can use
this "sensor" to identify hit pairs which capture the mRNA's
Shine-Dalgarno and AUG regions between them.
Cover Fraction is the fraction of mRNA "important region"
which the pair covers. The "important region" would usually be
defined as the mRNA region from the approximate Shine-Dalgarno location until
the AUG.
100% percent of the Bonus will be given to pairs which capture this
entire "mRNA important region" between them, while no bonus will be
given to pairs which do not capture any fraction of the "important
region" – and therefore won't be able influence translation initiation in
the most direct way of blocking the translation initiation site.
coverTargetStartPos
- Defines the mRNA position in which the "important" (usually SD-AUG)
region starts.
coverTargetEndPos
- Defines the mRNA position in which the "important" (usually SD till
AUG) region ends.
maxCoverBonus -
Maximal cover bonus value.
3. Ratio Bonus – Hits
Pair Ratio is defined as
the distance between the target gene positions of the two hits, divided by the
distance between the sRNA positions of the two hits (=The Distance between the
two hits on the mRNA divided by the distance between the two hits on the sRNA).
The Ratio is one of the most important elements of the SRTF program as
it is used to identify different binding models of sRNA molecules to target
gene mRNA molecules.
The following drawing displays the relation between the ratio value and
the sRNA-Target binding models. The only model verified in Lab is the
"Classic Double Kissing Complex" which is the binding model of the
oxyS-fhlA example (ratio value of 0.66).
All other models are theoretical. The drawing presents the mapping of
ratio value to the different models.
High ratio values are mapped to binding model of 2 hits in very close
proximity on the sRNA sequence, as Low ratio values are mapped to binding model
of 2 hits in very close proximity on the mRNA sequence. Moderate ratio values
are mapped to the classic double kissing complex – the oxyS-fhlA model.
Negative ratio values are mapped to CROSSED binding models.

The Ratio Bonus Function gives a bonus to pairs which their ratio is
within a desired range. Using Ratio Bonus, SRTF can give precedence to pairs
which implement a specific requested binding model.
Ratio Zones – The Ratio Bonus Function distinguished between 6
ratio zones. Each Zone is mapped to a different theoretical binding model.
Bonus will be given to pairs with ratio value which is found within one of the
requested zones.
![]() Example: "Ratio Zones" parameter of
"111000" would mean – give bonus to pairs with ratio values which are
found on the 3 first zones (the 3 negative zones ->requesting only crossed
models).
Example: "Ratio Zones" parameter of
"111000" would mean – give bonus to pairs with ratio values which are
found on the 3 first zones (the 3 negative zones ->requesting only crossed
models).
The Ratio Zones are defined according to two parameters (as described
in the drawing ahead) :
lowerRatioBound –
Lower ratio bound which would usually separate between 'mRNA Wrappers' and
'Classic Double Kissing Complex' models. Empirically set to 0.1 as default.
upperRatioBound -
Upper ratio bound which would usually
separate between 'sRNA Wrappers' and 'Classic Double Kissing Complex' models.
Empirically set to 1.5 as default.
maxRatioBonus -
Maximal ratio bonus value.
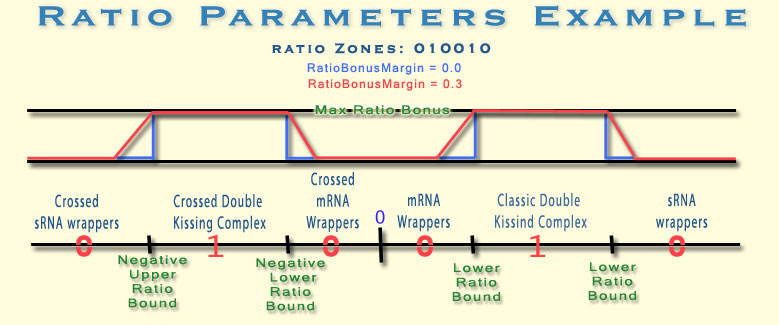
RatioBonusMargins
– This parameter is a fraction which determines the margin width of the ratio
bonus function. Value of 0 means sharp transitions from 0 points bonus to
maximal bonus, while value of 1 means slow and moderate decay of the bonus
function around the zone edges. Look at the example below.
enableTargetHitsOverlapping – Normally, SRTF would not allow
hit pairs which include two hits which overlap in their target gene windows
(the score of these pair is set to 0 and thus they will never be chosen as best
pair). Setting this Boolean parameter to 1 will enable overlapping of two hits
on the target gene.
enableSRnaHitsOverlapping - Normally, SRTF would not allow
hit pairs which include two hits which overlap in their sRNA windows (the score
of these pair is set to 0 and thus they will never be chosen as best pair).
Setting this Boolean parameter to 1 will enable overlapping of two hits on the
sRNA.
SRTF Web Site Help
The SRTF web
site enables you to execute analysis on pre-prepared data. SRTF data
infrastructure includes hits file for all currently known sRNA genes (Minimal
hit length: 5bp).
The Find Targets section is
the main website module, used to grade all genes according to a given set of
parameters.
Find
Targets Configuration Page
This screen is used to define the SRTF execution parameters.
sRNA name - Choose sRNA name for which you want to find targets.
Deep scan – SRTF
pre-prepared data includes two type of hit files.
Normal scan is faster, using hits file with minimal window size of 6, and
includes hits only at target position range -150 to 500. If the "Deep
scan" check box is checked, then SRTF will use hits file with minimal
window size of 5, and will include hits at target position range -150 to the
end of each gene.
Pre-defined
binding models – Each drawing presents a different (most are
theoretical …) binding model. Clicking on any of the model buttons will set the
execution parameters to a pre-defined parameters set, aim at identifying that
model.
Show
parameters –
Clicking the small '+' icon will open the parameters form where you can set any
of the parameters. You can first choose a pre-defined model by clicking one of
the buttons above, and then edit the parameters set to achieve the desired
result.
Show external
files - Here you can direct SRTF to use external files for
its analysis. The first list box enables you to choose a configuration file
name located on the server. In time, when more parameter sets are formed, more
configuration files can be put on the server. Next on this section you can
specify Hits file name, local configuration file (located on your computer) or
MFOLD connect (output of the MFOLD program for the sRNA you want to analyze).
After clicking the engage button, SRTF will start grading all genes
according to the parameters you have specified. SRTF execution time depends on
the following factors:
1.
sRNA
sequence length
2.
Range
of hits to be analyzed you have defined using the analyzeHitsFromPos and analyzeHitsUntilPos.
3.
Minimal
window size and target position hits range – Deep scan hit files will take much
longer time to run because there as their hit files contain many more hits.
4.
Server
workload.
If you do not wish to wait until the execution is over, you can either
copy the results link and review them later, or specify your e-mail address and
SRTF will send you an e-mail containing the results link.
Your execution results will remain on the server for several days.
Find
Targets results page –
The results page is divided to two parts: On the left you can see a listing of
all graded genes (including their rank number, name, grade and GI number which
is linked to the NCBI gene record). On the right part, you can see a graph
describing grades distribution and execution parameters. Clicking on a gene
name from the list will open the gene analysis page.
Gene
analysis page –
This page presents SRTF's analysis for a given gene.
The Hits table contains all hits included in the analysis – marked in pink are
the hits selected as best combination on phase 4.
Hit values and sub-scores are colored according to the slogan "The Pinker,
The better".
Above
the table you can see several grading details including ratio, distance and
cover values for the selected hits pair.
Clicking
on the "Graphic View" button will display a graphic representation of
the current gene.
Graphic
representation page –
This page enables you to view the MFOLD graphic output for the SRNA and current
gene. On the bottom you can see the sequences of both the sRNA and the current
gene, while the selected hits are marked in color.
SRTF express enables you to quickly grade a single gene according to
defined set of parameters. First define the execution parameters on the left
(including sRNA and gene names), hit the engage button and the gene analysis page for the requested gene should appear on the right.
Hits explorer enables you to query basic hit properties from a hits
file. The basic hit properties include target position, sRNA position, hit
length and hit energy. All of these parameters are basic and don't include any
assumption made by SRTF during the grading process of the "Find
Targets" and "SRTF Express" sections.
1. Altuvia,
S. Wagner, E. (2000) Proc. Natl. Acad. Sci. USA
97,9824-9826
2. Altuvia, S. Zhang, A. Argaman, L.
Tiwari, A. Storz, G. (1998)
EMBO Journal 17, 6069-6075
3. Argaman, L. Altuvia, S. (2000) J.
Mol. Biol. 300, 1101-1112
5. Chen,
S. Lesnik, E. Hall, T. Sampath,
R. Griffy, R, Ecker, D. Blyn, L. (2002) BioSystems
65, 157-177
6. Couzin, J. (2002) Science 298, 2296-2297
7. Delihas, N. Forst, S. (2001) J.
Mol. Biol. 313,1-12
8. Gottesman,
S. (2002) Genes & Development 16,2829-2842
9. Lease, R. Belfort, M (2000) Proc. Natl. Acad. Sci.
USA 97,9919-9924
10. Lease,
R. Belfort, M. (2000) Molecular Microbiology 38,667-672
12. Majdalani, N. Hernandez, D. Gottesman,
S. (2002) Molecular Microbiology 46,813-826
13. Nogueira,
T. Springer, M. (2000) Current Opinion in Microbiology 3,154-158
14. Rivas,
E. Klien, R. Jones, T. Eddy, S. (2001) Current
Biology 11, 1369-1373
15. Storz, G. (2002) Science 296,1260-1263
16. Tetart, F. Bouche, JP.
(1992) Molecular Microbiology 6,615-620
17. Wassarman, K (2002) Cell 109,141-144
19. Wassarman, K. Zhang, A. Storz, G.
(1999) Trends in Microbiology 7, 37-45
21.
Zuker, M. (1989) Science 244,48-51